唐铁山研究组合作发现肌萎缩侧索硬化症的致病新机制
细胞基因组DNA总是受到内源或外源环境中各种损伤因子的攻击,从而引起基因组DNA损伤。为维持基因组稳定性,生物体进化出了一种保护机制来监控DNA损伤并修复,这一机制即为DNA损伤应答。DNA损伤应答是一个复杂的信号传导网络系统,它能感知DNA损伤并将信号进行传递,进而引起一系列的应答反应,如激活细胞周期检验点、DNA修复、转录改变以及损伤过于严重时的细胞死亡等。
肌萎缩侧索硬化症(Amyotrophic lateral sclerosis,ALS),又叫渐冻症、葛雷克氏症,是一种慢性的运动神经元退行性疾病,伴随着过早的运动神经元的退行性病变和死亡,最终在临床上表现为致死性的肌肉麻痹无力和萎缩。ALS确切的发病机制至今尚不清楚,包括SOD1在内的多个基因突变与ALS发生密切相关。ALS的一个突出病理特征是脊髓运动神经元和胶质细胞胞质中形成含有tau (FTLD-tau)、TDP43、FUS、RBM45的包涵体。其中TDP43、FUS、RBM45都是RNA结合蛋白。RNA结合蛋白(RNA binding proteins, RBPs)在DNA损伤应答中的功能和机制研究是一个全新的研究领域。
本项研究发现RBM45可以被招募到DNA损伤位点,并且RBM45在损伤位点的招募依赖于PAR化(Poly(ADP-ribosyl)ation)但不依赖于RNA。RBM45缺失会导致异常的DNA损伤应答信号,降低同源重组(Homologous Recombination, HR)和非同源末端连接(Non-Homologous End Joining, NHEJ)效率,使得细胞对于离子辐照的敏感性明显增加。RBM45可以与去乙酰化蛋白HDAC1竞争性地结合FUS,而这种竞争性结合精细地调控了HDAC1到损伤位点的招募。家族性ALS相关的FUS突变体(FUS-R521C)与RBM45的亲和性显著增加,导致了HDAC1到损伤位点的招募缺陷和NHEJ修复效率的下降。考虑到NHEJ是终末分化的神经元修复双链断裂的首要途径,所以FUS突变-RBM45异常互作引发的HDAC1招募缺陷可导致神经元DNA损伤的累积,最终导致神经元功能异常和退行性病变。本项研究成果首次揭示了ALS相关蛋白RBM45在DNA损伤应答中的重要作用,同时对于阐释ALS的致病机理也有重要的意义。
这项研究工作以“RBM45 competes with HDAC1 for binding to FUS in response to DNA damage”为题于2017年11月14日在Nucleic Acid Research在线发表。本文第一作者为博士研究生巩娟娟、博士研究生黄敏以及博士后王凤丽,通讯作者为唐铁山研究员和北京基因组研究所郭彩霞研究员。本项目得到了科技部和基金委的资助。(文章链接)
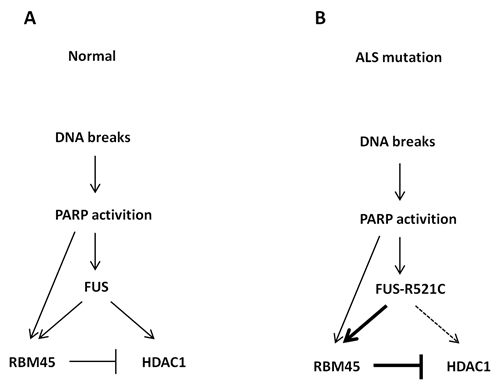
RBM45在正常情况 (A) 和ALS疾病时 (B) 参与DNA损伤应答过程的模式图